| Issue |
Knowl. Manag. Aquat. Ecosyst.
Number 425, 2024
Riparian ecology and management
|
|
|---|---|---|
| Article Number | 19 | |
| Number of page(s) | 13 | |
| DOI | https://doi.org/10.1051/kmae/2024014 | |
| Published online | 16 October 2024 | |
Research Paper
Population-genetics analysis of the brown trout broodstock in the “Panjica” hatchery (Serbia) and its conservation applications
1
Institute of Biology and Ecology, Faculty of Science, University of Kragujevac, Radoja Domanovića 12, 34000 Kragujevac, Serbia
2
Department of Animal Science, Biotechnical Faculty, University of Ljubljana, Jamnikarjeva 101, 1000 Ljubljana, Slovenia
3
Department of Zoology, National Museum of the Czech Republic, Václavské náměstí 68, 115 79 Prague 1, Czech Republic
4
Department of Ecology, Faculty of Science, Charles University, Viničná 7, 12844 Prague 2, Czech Republic
5
Department of Organisms and Ecosystems Research, National Institute of Biology, Večna pot 121, 1000 Ljubljana, Slovenia
6
Institute of Zoology, Faculty of Biology, University of Belgrade, Studentski trg 16, 11001 Belgrade, Serbia
* Corresponding author: This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
19
January
2024
Accepted:
26
August
2024
Abstract
Artificial propagation and stocking of brown trout is a standard practice in recreational fishery management. In recent decades, the importance of maintaining intraspecific diversity and protecting locally adapted lineages has been recognized for the species' long-term survival. The first step in selecting donors for stocking involves distinguishing native trout from non-native and introgressed individuals. The established method for discerning Atlantic hatchery strains from the wild populations involves genetic screening of individual diagnostic SNPs and microsatellite assignment tests. This study, using Serbia's Panjica hatchery as an example, illustrates the proper conduct of routine genetic screening for identifying suitable donors for supportive stocking. The broodstock and reference populations were screened using mtDNA control region, LDH nuclear gene, and 12 microsatellite loci to assess the origin, diversity, and inbreeding levels. The analysis revealed only moderate contamination with Atlantic trout and showed the regional origin of the Danubian genes – over 50% of the broodstock was composed of non-introgressed Danubian individuals tracing their origin to the Zapadna Morava River system. Additionally, the study highlighted a considerable discordance between LDH locus and microsatellites in identifying introgressed individuals, raising concerns about the sole reliance on LDH locus for the identification of Atlantic genetic origin in nuclear DNA.
Key words: molecular marker discordance / artificial propagation / Atlantic brown trout / Danubian brown trout / supplementation stocking
© T. Veličković et al., Published by EDP Sciences 2024
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License CC-BY-ND (https://creativecommons.org/licenses/by-nd/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. If you remix, transform, or build upon the material, you may not distribute the modified material.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License CC-BY-ND (https://creativecommons.org/licenses/by-nd/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. If you remix, transform, or build upon the material, you may not distribute the modified material.
1 Introduction
The brown trout (BT, Salmo trutta; here understood as all Salmo representatives excluding Salmo salar, S. ohridanus and S. obtusirostris) is characterized by the high levels of genetic diversity and complicated spatial patterns of genetic variations that indicate its complex evolutionary history (Bernatchez, 2001; Sanz, 2018; Veličković et al., 2023). Based on sequence variations of the mitochondrial (mt) DNA control region (CR), five major evolutionary lineages were described, Danubian (DA), Atlantic (AT), Adriatic (AD), Mediterranean (ME), and marmoratus (MA), followed by some additional, geographically more constrained lineages (Bernatchez et al., 1992; Bardakci et al., 2006; Vera et al., 2010; Snoj et al., 2011; Veličković et al., 2023). BT also exhibits high phenotypic diversity and may show considerable regional or even local differentiation, which plays an increasingly important and unavoidable role in conservation efforts (Fernández-Cebrián et al., 2014; Schmidt et al., 2017).
Due to the easy breeding and high yields, AT domesticated strains have spread greatly beyond their native range also including the Danube drainage and Mediterranean basin, where “AT genes” have become broadly introduced into native populations (García-Marín et al., 1991; Laikre et al., 1999; Poteaux et al., 1999; Jug et al., 2005; Splendiani et al., 2016; for details on BT phylogeography, see Bernatchez, 2001 and Veličković et al., 2023). AT domesticated strains originate from the western and north Atlantic river basins, where in the middle of the 19th century, massive production of BT started, and until recently these have been almost exclusive in hatcheries and for stocking (Berrebi et al., 2019, 2020, 2021).
Experience in BT management and related research has clearly shown that stocking of hatchery-reared AT brown trout is problematic due to competition and inter-breeding with native populations, which impair the adaptive potential and overall fitness of local individuals (Laikre et al., 1999; Dudgeon et al., 2006; Araki and Schmid, 2010; Caudron et al., 2011; Marić et al., 2022). In recent decades, the importance of maintaining intraspecific diversity and protecting locally adapted lineages has been recognized for the long-term survival of the species. To support the sustainable management of BT, conservation geneticists have conducted surveys of wild and hatchery populations (e.g., Laikre et al., 1999; Berrebi et al., 2019, 2021; Marić et al., 2022) to recognize trout lineages, identify introgressive hybridization, and estimate genetic diversity.
The first step in identifying suitable populations intended for stocking (donor populations) is to distinguish native populations, including native hatchery broodstocks, from non-native and introgressed populations. An established approach to distinguish Atlantic hatchery strains from the wild Danubian and Mediterranean populations relies upon the recognition of diagnostic SNPs on mtDNA CR (e.g., Bernatchez et al., 1992) and nuclear DNA (e.g., LDH gene; Hamilton et al., 1989; McMeel et al., 2001), and assignment tests, based on microsatellites loci. While mtDNA identifies the matrilineal descent of a population or individual, nuclear markers, in addition, provide information on introgressive hybridization, genetic admixture and other population parameters (Hansen et al., 2000, 2001; Chistiakov et al., 2005; Jones et al., 2009; Saura and Faria, 2011). While increasing number of BT studies are relying on high-throughput sequencing (HTS) and genome-wide datasets (e.g., RAD-seq, UCE; Hashemzadeh Segherloo et al., 2021) and, in some cases, sets of nuclear genes (Pustovrh et al., 2014; Snoj et al., 2021), these technologies are still fairly inaccessible for fisheries and wildlife managers. This is especially true for the Balkan countries that host high levels of BT variation but retain underdeveloped and insufficient HTS infrastructure. There, but also in other countries, individual diagnostic markers combined with microsatellites are still a valuable tool for determining the status of populations and individuals (Škraba Jurlina et al., 2018, 2020; Righi et al., 2023; Vera et al., 2023).
Serbia, in the central Balkans, hosts numerous non-introgressed native BT populations from two evolutionary lineages. The DA lineage is widespread, while the AD is found in the Aegean drainage in the southeast and in the Adriatic drainage in the south (Marić et al., 2006, 2022; Tošić et al., 2016; Simonović et al., 2017; Škraba Jurlina et al., 2020; Veličković et al., 2023). Stocking with BT is already a common practice in at least some parts of the country (Marić et al., 2022; Veličković et al., 2023). Consequently, non-native haplotypes, introduced from the Atlantic and Adriatic/Aegean watersheds, have been found in native Danubian BT in Serbia (Marić et al., 2006, 2017; Tošić et al., 2016; Simonović et al., 2017; Škraba Jurlina et al., 2020; Veličković et al., 2023). Additionally, introgressive hybridization with non-native BT from AD lineage (Aegean drainage in Serbia) has been observed in the native Danubian BT in the Vlasina Plateau (Southeast Serbia) (Marić et al., 2022).
According to the Law on Protection and Sustainable Use of Fish Stocks (Official Gazette of the Republic of Serbia, No. 128/2014 and 95/2018), stocking of fishing waters in Serbia must be conducted exclusively with native species. However, all BT phylogenetic lineages are treated as a single species under this law allowing stocking with material from virtually any origin. Additionally, the law does not mandate the preservation of the genetic diversity in local populations nor requires genetic testing of BT intended for stocking (Lenhardt et al., 2020). In Serbia, special legal authorization is required to rear and sell fish for stocking purposes (Official Gazette of the Republic of Serbia, No. 128/2014 and 95/2018). One of the largest hatcheries with the official authorization to produce stocking material is the Panjica hatchery (Hatchery “Braduljica” Ltd. Ivanjica) in southwestern Serbia. Despite its authorization to breed and sell BT throughout Serbia, the BT broodstock at this hatchery has not been thoroughly genetically tested.
To evaluate whether it produces suitable material for supplemental stocking of wild waters in accordance with conservation genetic guidelines, we used three different marker systems, i.e., mtDNA CR, microsatellite loci and LDH nuclear gene. This assessment aimed to determine the origin, genetic diversity, and inbreeding status of the broodstock. Using the Panjica hatchery as an example, we emphasize the importance of BT genetic testing and propose its implementation at the national level. This measure would help prevent the contamination of native populations with non-native hatchery genes, a problem that has already occurred and affected most of the BT distribution range.
2 Materials and methods
2.1 Hatchery broodstock origin
A preliminary CR characterization of the Panjica hatchery, conducted just a couple of years before undertaking the present study, revealed the predominant presence of non-native individuals. This finding aligned with the known history of fishery management at that time, indicating that the old broodstock was of mixed and partially unknown origin, containing both AT hatchery and native local BT (see Veličković et al., 2023). Driven by this analysis and efforts to introduce more sustainable stocking practices based on native broodstocks, hatchery managers restored the old broodstock with BT sourced locally from the Zapadna Morava River system, primarily from a non-introgressed DA population in the Panjica River. However, a smaller proportion of donors came from a few other nearby sources, some of which were introgressed with non-native BT (see Veličković et al., 2023 for mtDNA CR screening of brown trout in Serbia). A few years after the broodstock was replenished, we genotyped and analyzed it along with reference populations to assess its status, as demonstrated in the research presented here. This analysis was intended to guide the subsequent selection processes for producing a new native DA hatchery broodstock (novel broodstock).
According to the information provided by the Panjica hatchery staff, the novel broodstock was established in 2021 using solely native DA individuals (Group 1; see Results). This broodstock was kept in separate tanks from the other hatchery population. For the first spawning of the novel broodstock, the hatchery employed broodstock selection based on the condition characteristics of individuals followed by random pairing.
2.2 Sampling and DNA isolation
Sampling was carried out in July 2019 at the Panjica hatchery (Dobrače, southwestern Serbia; Tab. 1). One hundred and forty-one brown trout were fin-clipped and subcutaneously implanted with a microchip (FDX-B transponder, Virbac). These individuals were not from the same cohort and were kept in the hatchery for a couple of years prior to the analysis. According to the results (see Sect. 3.1. for further information), Panjica hatchery samples were divided into three groups: Group 1, “purest” DA individuals without detected introgression; Group 2, “hybridized” individuals; and Group 3, individuals showing “pure” AT origin. Additionally, in 2022, 20 native DA offsprings of the novel broodstock were sampled and analyzed to assess the effectiveness of the selection recommended in this work (Tab. 1).
Total DNA was isolated from the fin clips (preserved in ethanol) using the Tissue Genomic DNA Mini Kit (Geneaid Biotech Ltd., Taiwan). The study also included Atlantic brown trout reared in the Braduljica hatchery (Ivanjica, southwestern Serbia), as well as previously studied reference population from the Panjica River (Serbia), a population of non-introgressed native Danubian origin (Veličković et al., 2023) and three populations of Atlantic origin from Denmark (hatchery population), Austria (Lake Attersee), and Bosnia and Herzegovina (Trebišnjica River; Marić et al., 2022; Veličković, 2023), bringing the total number of samples to 216 (Tab. 1).
Sampling sites, number of individuals sampled (N), drainage/origin, and geographic coordinates of Panjica hatchery and reference populations. Panjica hatchery groups: (Group 1) native stock, (Group 2) admixed stock, (Group 3) introduced stock.
2.3 Mitochondrial DNA
The complete mtDNA CR (ca. 1100 bp) was amplified with primers LRBT-25 and LRBT-1195 (Uiblein et al., 2001) under the conditions described in Marić et al. (2022). DNA sequencing in both directions using the same primer pair was performed by Macrogen Europe (Amsterdam, Nederland). Sequences were edited and aligned using Chromas Lite v. 2.6.5 (http://www.technelysium.com.au/chromas.html; Technelysium Pty Ltd, Australia). BLASTn (Altschul et al., 1990) was used to compare the sequences with those available in NCBI GenBank (Sayers et al., 2022). The haplotype network was constructed in PopART v. 1.7 software (Leigh and Bryant, 2015) using the phylogenetic median-joining (MJ) network algorithm (Bandelt et al., 1999). In addition to the CR sequences from the present study, previously published haplotypes of the Danubian (DA), Adriatic (AD), Mediterranean (ME), marmoratus (MA), Atlantic (AT), and Duero (DU) evolutionary lineages were also used (see Tab. S1 for the haplotypes details). For CR sequence variation, pairwise differentiation (ФST) among populations was estimated using Arlequin v. 3.5 (Excoffier and Lischer, 2010), based on the Tamura–Nei (TN93; Tamura and Nei, 1993) nucleotide substitution model. The statistical significance was based on permutation tests of 10,000 replications.
For 20 offspring of the novel broodstock, PCR amplification and RFLP analysis of the CR amplicons using the SatI endonuclease was performed to discern the DA and AT lineages, following the procedure described in Marić et al. (2010).
2.4 Partial LDH-C* locus
PCR amplification and RFLP analysis of the nuclear LDH gene (428 bp) using the BseLI endonuclease was performed as in Marić et al. (2010). We used this marker to distinguish Danubian BT, carrying the ancestral allele 100, from the hatchery-reared Atlantic BT, carrying the derived allele 90 (McMeel et al., 2001; Fig. S1). The GDA program (Lewis and Zaykin, 2001) was used to calculate genetic diversity, including the mean number of alleles per locus (A), observed (HO) and expected heterozygosity (HE), and the inbreeding coefficient (FIS).
2.5 Microsatellite DNA
Twelve microsatellite loci were included in the analysis using primers for PCR amplification and multiplex PCR conditions, as in Lerceteau-Köhler and Weiss (2006). Amplicons were separated on an ABI 3500 GA capillary sequencer using the GeneScan 500 Rox Size Standard (Applied Biosystems). Fragment lengths were determined using GeneMapper® v. 5.1 software (Applied Biosystems). Micro-Checker v. 2.2.3 (Van Oosterhout et al., 2004) and MicroDrop v. 1.01 (Wang et al., 2012) were used to check for the presence of null alleles and to test for allelic dropout.
All downstream analyses except comparison of genetic diversities and STRUCTURE analysis were performed for microsatellite and LDH-C* data together. A general picture of the BT diversity was described through a Factorial Correspondence Analysis (FCA) as implemented in Genetix v. 4.04 (Belkhir et al., 1996–2004). The same software was used to calculate gene diversity (heterozygosity) and the average number of alleles per locus per population/group. Allelic richness was estimated by rarefaction analysis, using ADZE (Szpiech et al., 2008) to assess whether sampling effort was sufficient to capture genetic diversity. Comparisons of obtained genetic diversity values (Ho, He, A, and Ar) between Panjica River BT and three groups within Panjica hatchery broodstock (see Results) were further evaluated using a Mann–Whitney U-test implemented in the PAST v. 4.16 (Hammer et al., 2001). Deviations from the HWE were examined for each locus/population combination as well as across all loci and samples with Fisher's method as employed in Genepop v. 4.8.3 (Rousset, 2008) using 10,000 dememorization steps, 100 batches and 10,000 iterations per batch. The FIS values (inbreeding coefficient; within and over loci/populations) along with the 95% confidence intervals (95% CI) and pairwise coefficients of population differentiation (FST, Weir and Cockerham, 1984) were evaluated using the hierfstat library (de Meeûs and Goudet, 2007; Goudet and Jombart, 2022) in R CRAN (R Core Team, 2022). Tests for linkage disequilibrium (LD) and its significance were done in Genepop using the same Markov chain parameters as above.
The genetic structure was assessed using STRUCTURE v. 2.3.4 software (Pritchard et al., 2000). MCMC (Markov Chain Monte Carlo) was run for 1,000,000 replicates after a burn-in of 250,000 generations. The number of clusters, K, in the dataset was explored from K = 1 to K = 8, and seven replicates were performed for each value. The ΔK method (Evanno et al., 2005) was applied to estimate the most likely K. The results of STRUCTURE were compared with those of NewHybrids v. 2.0+ Developmental (Anderson and Thompson, 2002; Anderson, 2008), which was used to calculate posterior probabilities for each individual to be assigned to the following classes: DA lineage, AT lineage, first generation hybrids (F1), second generation hybrids (F2) and backcrosses with each parental lineage (BC-DA, BC-AT). This program was run with uniform and Jeffrey uniform priors and with a burn-in period of 100,000 steps, followed by 300,000 iterations to estimate individual posterior probabilities.
Potential full sibling groups of the selected individuals (Group 1; see Results) were identified using the pairwise-likelihood/full-likelihood score combined method (FPLS) as implemented in COLONY v. 2.0.6.5 (Jones and Wang, 2010).
3 Results
3.1 Sorting Panjica broodstock brown trout based on the Danubian and Atlantic genetic proportion
After the initial analyses (Figs. 1, 2, and Tab. 2), the genetic test based on CR, LDH, and microsatellites (the details for each marker system are provided in Sect. 3.2) placed the individuals of the Panjica broodstock into three groups (Tabs. 1, 2, and 3, Fig. 3):
Group 1, 70 individuals possessing DA CR haplotype, 100/100 LDH genotype and microsatellite-based Danubian genetic proportion >0.99. We considered these individuals to be native.
Group 2, 66 individuals showed introgression on at least one of the three marker systems used.
Group 3, five individuals with AT CR haplotype, 90/90 LDH genotype and microsatellite-based Atlantic genetic proportion > 0.99. We assumed these individuals to be Atlantic hatchery-reared.
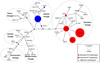 |
Fig. 1 Median-joining network of brown trout mtDNA CR haplotypes. Previously described haplotypes that were not detected in this study are represented by grey circles of the same size. For haplotypes detected in the Panjica hatchery broodstock, the size of the cirlces reflects the frequency of each haplotype, and they are color coded (red – DA, blue – AT lineage). Mutations are represented by slashes crossed with the network branches. The black circle indicates an extinct, ancestral or unsampled haplotype. |
 |
Fig. 2 Estimated population structure as inferred by STRUCTURE analysis of microsatellite DNA data. White lines separate sampling sites. The most likely K for the analyzed samples is based on the ΔK method; no further structures were detected after the first step and within the excluded clusters. The population structure of the Panjica hatchery broodstock is magnified and shown between the arrows, with each column representing one individual (the number corresponds to the nominal number of individuals from Tab. S4). The red color illustrates the genetic proportion of the DA and the blue of the AT. |
Frequency of mtDNA CR haplotypes, LDH alleles and genotypes, and LDH and microsatellite DNA genetic diversity of Panjica hatchery broodstock.
Frequency of mtDNA haplotypes, LDH genotypes, and microsatellite DNA gene diversity for three groups of Panjica hatchery broodstock and reference populations. Panjica hatchery groups: (Group 1) native stock characterized by DA mtDNA CR haplotype, 100/100 genotype and a very high proportion of DA alleles (> 0.99), (Group 2) admixed stock where discrepancies on different markers were detected, (Group 3) introduced stock characterized by AT mtDNA CR haplotype, 90/90 genotype and a very high proportion of AT alleles (> 0.99).
 |
Fig. 3 mtDNA haplotype, LDH genotype, and proportion of Danubian alleles of microsatellite DNA of each individual from the Panjica hatchery broodstock including the novel broodstock. The red color illustrates the genetic proportion of the DA and the blue of the AT. Each column represents one individual on three different markers (the number of individuals corresponds to the nominal number from Tab. S4). The group assigned to the individual corresponds to Table S4. |
3.2 Results for each marker system
3.2.1 Mitochondrial DNA
Sequencing of the mtDNA CR provided readable sequences of 992 bp. Seven CR haplotypes were detected in the Panjica hatchery broodstock, four of which belong to the DA lineage and three to the AT lineage (Tab. 2). Of the 141 individuals analyzed, 108 (76.6%) had DA haplotypes, while the remaining 33 (23.4%) had AT haplotypes. The most common haplotype was Da28, followed by ATcs3 and Da1c and Da1a, which were detected in similar frequencies, while other haplotypes were detected in only one or two individuals (Tab. 2, Fig. 1). Da28 and Da1a were also detected in the Panjica River (Tab. 3). The remaining AT haplotypes found in the Panjica hatchery broodstock are all common hatchery haplotypes and were also observed in the reference AT populations (Tab. 3). Haplotypes detected in the Panjica hatchery broodstock clearly grouped within DA and AT lineages in the MJ network (Fig. 1). Here, the central DA haplotype was Da1a, and the three other haplotypes from the present study (Da28, Da1c, and Da2a) were separated from it by one mutational step. Within the AT lineage, ATcs3 was centrally positioned (Fig. 1). The PCR-RFLP analysis of the mtDNA CR showed that all 20 individuals of the novel broodstock belonged to the DA lineage (Tab. 3). Pairwise ФST values were in general low and non-significant among the reference AT populations, including Panjica hatchery Group 3, and intermediate to high and statistically significant among the Panjica River BT and reference AT populations and Panjica hatchery Group 1 and reference AT populations (Tab. 4).
Pairwise ФST (mtDNa CR; above) and FST values (nuclear loci – combined microsatellites and LDH gene; bellow) with significance values, offsprings of the novel broodstock where genotyped at mtDNA CR using only RFLP approach that discriminates between DA and AT lineage.
3.2.2 Partial LDH-C* locus
In the Panjica hatchery, allele 100 was predominant (74.5%), while the frequency of allele 90 was 25.5%. The frequencies of the 90/90, 90/100 and 100/100 genotypes were 8.5%, 34.0% and 57.5%, respectively. The LDH genetic diversity values indicated a mixed origin of the hatchery individuals (Tab. 2). All individuals from the Panjica River and the novel broodstock exhibited the genotype 100/100, while all reference AT populations showed the genotype 90/90 (Tab. 3).
3.2.3 Microsatellite DNA
No null alleles were detected, and all populations/groups except Group 2, the novel broodstock and the Attersee Lake were in HWE (Tab. 3). Considering Bonferroni correction, only Group 2 did not conform to HWE expectations. Group 2 was characterized by the highest levels of LD – significant LD (initial α = 0.01; considering Bonferroni correction) was detected at 17 loci pairs there and only at two loci pairs in Group 1. LD was also detected at a single locus pair in the offsprings of the novel broodstock but not in any of the reference populations.
Genetic diversity parameters (Ho, He, A, Ar) were lowest in the Panjica wild population and considerably higher in all hatchery populations (Tab. 3). The FIS values were (very) low in the entire data set and negative in most populations (−0.081–0.007), indicating an excess of heterozygotes. Panjica hatchery FIS values were considerably higher (less negative) when analyzed as a single group (−0.014) than when sorted into three groups (lower than −0.046). Genetic diversity values (Ho, He, A, and Ar) did not show significant differences between the Panjica BT and three Panjica hatchery groups, as indicated by Mann-Whitney U-tests (P > 0.01 for all diversity estimators; Tab. S2). The exception was observed between the Panjica BT and Group 3 (introduced stock), where statistically significant differences were noted in both Ho and Ar values (Mann-Whitney U: 23.5 vs. 27, Z = −2.77128, P = 0.0056; Mann-Whitney U: 22 vs. 27, Z = −2.85788, P = 0.00424, respectively) (Tab. S2). When comparing pairwise FST values between the three Panjica hatchery groups (see Sect. 3.1) and reference populations, they were lowest when comparing Group 1 to the DA reference population (Panjica River) and Group 2 and 3 to one of the AT reference populations (0.228, 0.109, and 0.006, respectively) and highest when switching the references (0.273, 0.222, and 0.435). Pairwise comparisons between Group 3 and reference AT population(s) were among the lowest in overall, while the highest values were observed when comparing the DA and AT reference populations (0.381–0.424; Tab. 4).
The results of the STRUCTURE analysis matched the patterns observed from FCA (Fig. S2), with the most probable value of K = 2 (Tab. S3) revealed groups corresponding to Danubian and Atlantic ancestry. The first group (Fig. 2, red) was the only one present in the Panjica BT and is hereafter referred to as the Danubian cluster, while the second group (Fig. 2, blue) was the only one present in the Braduljica hatchery, Danish hatchery, Lake Attersee, and Trebišnjica population, hereafter referred to as the Atlantic cluster (Fig. 2, Tab. S3). Individuals from the Panjica hatchery broodstock were distributed between both groups, with most (104 individuals, 73.8% of the broodstock) having no AT or very little introgression with AT (> 0.99 of the Danubian alleles) – one individual had an estimated proportion of 0.97 of DA genes, and one of 0.86 (Figs. 2 and 3, Tab. S4). All non-introgressed individuals carried the native DA haplotype; however, among them, 32 were heterozygous (90/100) for LDH alleles and three were homozygous for the hatchery LDH 90 allele. Of the remaining 35 individuals, nine had introgression levels of 0.15–0.49, all carrying AT haplotypes, while all LDH allelic combinations, including 100/100, were observed within this introgressed group. The remaining 26 individuals belonged to the Atlantic group with the Danubian genetic proportion under 0.07. The other marker systems (i.e., mtDNA CR and LDH; Fig. 3) also showed that these individuals were predominantly of Atlantic origin. The overall average genetic proportion of DA within the broodstock was 0.75. The NewHybrids analysis showed a pattern similar to that obtained with STRUCTURE (Fig. 4). It confirmed that nine previously identified admixed genotypes were likely to be F1 hybrids and that the DA origin of the 104 pure or nearly pure individuals was correctly estimated. However, among the remaining 26 individuals previously identified as belonging to the AT group, this analysis assigned 17 to F1 hybrids and only nine to the AT group or backcrosses with the latter. All 20 offspring from the novel broodstock were placed in the Danubian cluster according to both STRUCTURE and NewHybrid analysis (Figs. 2 and 4).
Parentage analysis was performed for Danubian Group 1 and identified 11 full-sibling families. Each full-sibling family consisted of two individuals. Seven full-sibling families had Prob (Inc.) values greater than 0.5 and Prob (Exc.) less than 0.5 (Tab. S5).
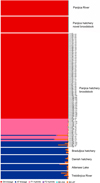 |
Fig. 4 NewHybrids analyses using uniform priors based on microsatellite data. The color illustrates the estimated posterior probabilities that an individual belongs to one genotype frequency categories: DA lineage (red), AT lineage (blue), F1 hybrids (pink), F2 hybrids (violet), backcrosses with DA (turquoise), and crosses with AT (orange). |
4 Discussion
4.1 Concordance among molecular markers
The three types of markers used in the present study to analyze BT broodstock in the Panjica hatchery varied from each other in terms of mutation rates and mode of inheritance, thus representing independent determination systems. Despite these differences, the markers produced very similar results – at least half of the broodstock (i.e., Group 1 in Fig. 3) consisted of apparently non-introgressed Danubian individuals, suggesting a good potential for future propagating of native BT in Serbia. However, it is important to clarify that the term 'non-introgressed', strictly speaking, refers only to the genome covered by the diagnostic markers. If we consider that the population is subject to natural processes, we must also acknowledge the possibility that the entire tested population is introgressed, meaning that some hybrids might not be detected with the available markers. In addition, simulations have shown that a few generations of random mating between the native and hatchery BT reduces the detection of released hatchery fish using microsatellites (Sanz et al., 2009). On the other hand, the incidence of false negatives (i.e., false identification of introgressed individuals as non-introgressed) in a random mating hybrid swarm decreases considerably with increasing number of diagnostic loci. For example, there is only ca. 0.1% probability for a false negative observation given a 10% introgression level using six diagnostic nuclear markers (see Allendorf et al., 2001). Given the number of microsatellite markers (12 markers) we used in the present study to determine the Atlantic genetic proportion, and especially considering the high power of microsatellite loci to distinguish hybrids, there is a high probability that the entire Group 1 consists of genuinely non-introgressed or minimally introgressed individuals.
High genetic diversity, previously documented for brown trout broodstocks across Europe (Bohling et al., 2016), was also observed in the Panjica hatchery in overall. The increase of genetic diversity is primarily due to the genetic mixing of native breeders with imported Atlantic brown trout (Berrebi et al., 2017) and, to a lesser extent, the use of different local sources in forming the broodstock in the years preceding the analysis (see Sect. 2.1). Besides the Panjica River, smaller proportions of donors also originated from other nearby sources (all from the Zapadna Morava River system), some of which are at least partially introgressed with non-native trout (also see Veličković et al., 2023).
In the introgressed individuals (Group 2; Fig. 3, Tab. S4), it is important to note that in more than half of these individuals, Atlantic introgression is only attributed to the presence of allele LDH 90, which mostly occurs in a heterozygous state with allele LDH 100. This represents a discrepancy compared to the introgression detected with microsatellites. To our knowledge, such discordance has not yet been reported for hatchery populations. A similar mismatch was observed in wild marble trout hybrids in the Soča River system (Slovenia), where analysis of the LDH locus showed much higher Danubian BT introgression compared to microsatellites (Berrebi et al., 2017). This was explained as an ancient natural immigration of the Danubian BT followed by saturation of the microsatellite phylogenetic signal. However, these two cases are difficult to compare. The formation of Panjica broodstock is a recent, anthropogenically caused event, whereas the Soča marble trout case is diachronic and, therefore, subject to other evolutionary drivers.
On the other hand, a recent study from the Iberian Peninsula compared estimates of the genetic proportion of hatchery BT in wild populations for the LDH gene, microsatellite markers (five loci) and a SNP panel (19 loci). This study showed high concordance between the markers, especially between the SNP panel and LDH. However, this evaluation was only performed at the population level and not at the individual level (Casanova et al., 2022). The discrepancy in Panjica introgression might, in theory, also be a result from ancestral polymorphism. There have been very few studies on Danubian populations, particularly in the Balkans, focused on LDH typing. Therefore, it is possible that native allele 90 is present in unexplored BT populations, making it difficult to deny its presence.
The introgressive discrepancy could also be explained as a theoretically possible result of the positive selection of the 90 allele under fish farming conditions. LDH may not act as a neutral marker as LDH-C* allozymes have different properties (Henry and Ferguson, 1985) that may be of selective value depending on environmental conditions (Horreo et al., 2015). Coding genes, unlike hypervariable neutral markers such as microsatellite loci, have a slower evolution rate and are subject to selection, making them less prone to genetic saturation, bottleneck effects, and random genetic drift after broodstock formation in hatcheries (Ward, 2000; Berrebi et al., 2017). In a shielded environment, such as hatchery conditions, individuals with the allele 90 may outperform the others, as demonstrated for BT stocked in the Narcea system in Spain (Horreo et al., 2015). This raises the question: why is there no trace of microsatellite introgression in LDH heterozygotes from the Panjica broodstock, given the neutrality of microsatellite markers? Additionally, why did not selection eliminate more than half of the individuals that are 100/100 homozygotes? According to the Panjica hatchery staff the hatchery trout prior to the formation of the novel broodstock followed random pairing, while the selection of the spawners for the novel broodstock was made based on the condition characteristics of individuals. This raises concerns about whether this method may have influenced the genetic structure in ways that were not anticipated and might have contributed to the divergence of the novel broodstock. In our opinion, the next most likely explanation involves anthropogenic factors, such as a recent, rather limited introduction of the Atlantic BT (Group 3 in Fig. 3) and various combinations of parental crosses and backcrosses with their offspring. These factors, in conjunction with the hatchery's crossbreeding practices, can result in unusual genetic signatures that are unlikely to occur naturally. Specifically, admixture LD between loci that are otherwise unlinked suggests recent admixture (over a few generations) and/or assortative mating, both of which argue against a hybrid swarm scenario.
Introgression discrepancies between mtDNA and nuclear data were also observed at the individual level (Tab. S4, Fig. 3). However, such discrepancies are common as maternally inherited mtDNA is fixed much faster (i.e., four times faster) than nuclear DNA markers; also, bottlenecks or specific (peculiar) crossing schemes that are frequent companions of fish manipulations in hatcheries cannot be excluded.
4.2 Importance of genetic characterization of brown trout hatchery broodstocks for fisheries management guidelines
This study provides insights into the origin and composition of the studied Panjica hatchery population and offers a practical framework for addressing similar management issues. Results presented here have already been directly incorporated into the management practices of the Panjica hatchery. Only breeders from the native Danubian genetic group (Group 1; individuals listed in Tab. S4) were selected, resulting in the creation of a ‘novel broodstock’. To avoid the deleterious effects of inbreeding (Hansen and Jensen, 2005; McLean et al., 2008; Vera et al., 2010), a sibling analysis was conducted to identify and exclude full siblings from the novel broodstock (Tab. S5). Consequently, 63 individuals met all criteria for final inclusion in the novel broodstock to be used for propagating native DA BT (Tab. S4). Considering the genetic composition of Panjica hatchery broodstock, individuals from this hatchery are suitable for stocking parts of the Zapadna Morava River system. Additionally, establishing other regional DA broodstocks for other parts of the Danube basin is recommended. In many countries, different evolutionary BT lineages and haplogroups co-exist naturally (e.g., Bernatchez, 2001; Jug et al., 2005; Jadan et al., 2015; Splendiani et al., 2016; Marić et al., 2022; Veličković et al., 2023). Therefore, it is of utmost importance that hatchery-reared BT used for supplemental stocking belong to the same haplogroups or evolutionary lineage and even carry the same haplotypes as the recipient wild population. Furthermore, local differentiation in nuclear genes must also be considered (see Sect. 4.1). Before establishing new broodstocks in Serbia or other countries, both source and potential sink populations should undergo a thorough genetic screening that is at least as comprehensive as the one presented in this study.
Implementing a native stocking program that maintains local genetically distinct populations also at intra-drainage levels is challenging due to limited infrastructure, the costs associated with genetic screening, and the high economic burden of maintaining multiple native stocks. However, neglecting genetic differences between populations in the management of BT may prove even more damaging in environmental, financial and socio-economic terms. To overcome this challenge, Serbia and many other countries (e.g., Croatia; Kanjuh, 2023) should improve their national legislation to ensure that genetic differences between populations are considered for the effective conservation of BT.
Based on the experience gained in this work, we make recommendations for the establishment of a native hatchery broodstock:
Emphasize population genetic analysis: To achieve the genetic goals associated with nativeness and to ensure the genetic integrity of stocked populations, population genetic analysis during stock establishment is crucial.
Mark and genotype wild native donors: When selecting wild native donors for artificial breeding, mark and genotype them prior to propagation. This approach helps to avoid the introduction of introgressed local donors, as was observed in the formation of the current Panjica broodstock.
Frequent refreshment of broodstock: Regularly refresh the broodstock with spawners to mitigate the problems with domestication (Frankham, 2008; Williams and Hoffman, 2009). This practice helps maintain genetic diversity and reduce the risk of inbreeding.
Utilize different native local sources: Regular refreshment with different native local sources should also prevent homogenization of populations across management units, which can occur even if native sources are used (also see Fernández-Cebrián et al., 2014). However, when a population genetic structure exists, this practice can lead to homogenization of wild populations and the loss of their local adaptations. Therefore, a prior genetic analysis is imperative to retain the highest genetic diversity present in wild populations.
By following these recommendations, hatcheries can better maintain the genetic integrity of brown trout populations and support the conservation of native genetic lineages.
Supplementary material
Fig. S1. Restriction of the partial fragment of the LDH gene (428 bp) with the BseLI enzyme. Allele 100 of the DA lineage remains uncut, whereas allele 90 of the AT lineage was cut into two fragments (353 and 75 bp; some fragments of 75 bp were of very low concentration and hardly visible on the gel).
Fig. S2. Two-dimensional plot of Factorial Correspondence Analysis (FCA) for the entire sample-set based on 12 microsatellite loci.
Table S1. GenBank accession numbers of sequences detected in the study along with the reference sequences used for construction of the Median-joining network. Sequences detected in the Panjica hatchery are in bold.
Table S2. Statistical differences in genetic diversity values (Mann-Whitney U tests: P < 0.01) between Panjica River and three Panjica hatchery groups (1 – native, 2 – admixed, 3 – introduced stock). Abbreviations: Ho – observed heterozygosity; He – expected heterozygosity; A – average number of alleles per locus; Ar – allelic richness.
Table S3. Estimation of the number of genetic clusters from STRUCTURE runs using the ΔK method. Selected number of clusters is highlighted.
Table S4. List of mtDNA haplotype, LDH genotype, and proportion of Danubian alleles of microsatellite DNA of each individual from the Panjica hatchery broodstock. The last column indicates the group assigned to the individual (1 – native, 2 – admixed, 3 – introduced stock). Individuals assigned as native according to all markers (DA mtDNA haplotype, 100/100 genotype, and > 0.99 Danubian alleles of microsatellite DNA) are printed. Individuals highlighted in gray are those that were to be remain in the final DA native broodstock, after the analysis of parentage assessment (Tab. S5).
Table S5. Full-sib families belonging to Group 1 (native) Danubian Panjica hatchery individuals. Each row represents one family. Full Sibhip index – the family index the 1st column; Prob (Inc.) – inclusive probability for this family; Prob (Exc.) exclusive probability for this family; Ind. 1 – sample number of the first offspring of the family, Ind. 2 – sample number of the second offspring of the family. Rows containing Prob (Inc.) higher than 0.500 and Prob (Exc.) lower than 0.500 are highlighted gray, § marks the family member suggested to be excluded from the final DA native broodstock.
Access Supplementary MaterialAcknowledgements
We thank the reviewers and editor for their constructive feedback and valuable suggestions, which helped us to improve the quality of the manuscript.
Funding
TV, VS, and SM were supported by the Serbian Ministry of Science, Technological Development and Innovation (TV and VS, Grant No. 451-03-65/2024-03/200122; SM, Grant No. 451-03-65/2024-03/200178). The study was co-financed by the Slovenian Research Agency (DS, Research programme No. P1-0255 and Research project No. J1-3015; AS and JB, Research program No. P4-0220). DS was additionally supported by the Ministry of Education, Science and Sport of Slovenia and the European Regional Development Fund (project RI-SI-2 LifeWatch). RŠ was supported by the Ministry of Culture of the Czech Republic (DKRVO 2024–2028/6. I.a, National Museum, 00023272), JV by institutional resources of the Ministry of Education, Youth and Sports of the Czech Republic. The authors express their gratitude to Milan Lugić from the Hatchery “Braduljica” Ltd. Ivanjica for his irrepressible support during the realization of this study and to anonymous reviewers and the editor for their constructive comments.
References
- Allendorf FW, Leary RF, Spruell P, Wenburg JK. 2001. The problems with hybrids: setting conservation guidelines. Trends Ecol Evol 16: 613–622. [CrossRef] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215: 403–410. [CrossRef] [PubMed] [Google Scholar]
- Anderson EC. 2008. Bayesian inference of species hybrids using multilocus dominant genetic markers. Philos Trans R Soc Lond B Biol Sci 363: 2841–2850. [CrossRef] [PubMed] [Google Scholar]
- Anderson EC, Thompson EA. 2002. A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160: 1217–1229. [CrossRef] [PubMed] [Google Scholar]
- Araki H, Schmid C. 2010. Is hatchery stocking a help or harm? Evidence, limitations and future directions in ecological and genetic surveys. Aquaculture 308: S2–S11. [CrossRef] [Google Scholar]
- Bandelt H, Forster P, Röhl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48. [CrossRef] [PubMed] [Google Scholar]
- Bardakci F, Degerli N, Ozdemir O, Basibuyuk HH. 2006. Phylogeography of the Turkish brown trout Salmo trutta L.: mitochondrial DNA PCR-RFLP variation. J Fish Biol 68: 36–55. [CrossRef] [Google Scholar]
- Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F. 1996–2004. GENETIX 4.05. logiciel sous Windows TM pour la genetique des populations. Laboratoire Genome, Populations, Interactions, CNRS UMR 5000. Universite de Montpellier II. Montpellier (France). [Google Scholar]
- Bernatchez L. 2001. The evolutionary history of brown trout (Salmo trutta L.) inferred from phylogeographic, nested clade, and mismatch analyses of mitochondrial DNA variation. Evolution 55: 351–379. [Google Scholar]
- Bernatchez L, Guyomard R, Bonhomme F. 1992. DNA sequence variation of the mitochondrial control region among geographically and morphologically remote European brown trout Salmo trutta populations. Mol Ecol 1: 161–173. [CrossRef] [PubMed] [Google Scholar]
- Berrebi P, Jesenšek D, Crivelli AJ. 2017. Natural and domestic introgressions in the marble trout population of Soča River (Slovenia). Hydrobiologia 785: 277–291. [Google Scholar]
- Berrebi P, Barucchi VC, Splendiani A, Muracciole S, Sabatini A, Palmas F, Tougard C, Arculeo M, Marić S. 2019. Brown trout (Salmo trutta L.) high genetic diversity around the Tyrrhenian Sea as revealed by nuclear and mitochondrial markers. Hydrobiologia 826: 209–231. [CrossRef] [Google Scholar]
- Berrebi P, Marić S, Snoj A, Hasegawa K. 2020. Brown trout in Japan − introduction history, distribution and genetic structure. Knowl Manag Aquat Ecosyst 421: 18. [CrossRef] [EDP Sciences] [Google Scholar]
- Berrebi P, Horvath Á, Splendiani A, Palm S, Bernas R. 2021. Genetic diversity of domestic brown trout stocks in Europe. Aquaculture 544: 737043. [CrossRef] [Google Scholar]
- Bohling J, Haffray P, Berrebi P. 2016. Genetic diversity and population structure of domestic brown trout (Salmo trutta) in France. Aquaculture 462: 1–9. [CrossRef] [Google Scholar]
- Caudron A, Champigneulle A, Guyomard R, Largiader CR. 2011. Assessment of three strategies practiced by fishery managers for restoring native brown trout (Salmo trutta) populations in Northern French Alpine Streams. Ecol Freshw Fish 20: 478–491. [CrossRef] [Google Scholar]
- Casanova A, Heras S, Abras A, Roldán MI, Bouza C, Vera M, García-Marín JL, Martínez P. 2022. Genomic hatchery introgression in brown trout (Salmo trutta L.): development of a diagnostic SNP panel for monitoring the impacted mediterranean rivers. Genes 13: 255. [CrossRef] [PubMed] [Google Scholar]
- Chistiakov DA, Hellemans B, Volckaert FA. 2005. Microsatellites and their genomic distribution, evolution, function and applications: a review with special reference to fish genetics. Aquaculture 255: 129. [Google Scholar]
- Dudgeon D, Arthington A, Gessner M, Kawabata Z, Knowler D, Lévêque C, Naiman R, Prieur-Richard A, Soto D, Stiassny M, Sullivan C. 2006. Freshwater biodiversity: importance, threats, status and conservation challenges. Biol Rev 81: 163–182. [CrossRef] [PubMed] [Google Scholar]
- de Meeûs T, Goudet J. 2007. A step-by-step tutorial to use HierFstat to analyse populations hierarchically structured at multiple levels. Infect Genet Evol 7: 731–735. [CrossRef] [PubMed] [Google Scholar]
- Evanno G, Regnaut S, Goudet J. 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620. [CrossRef] [PubMed] [Google Scholar]
- Excoffier L, Lischer HEL. 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567. [Google Scholar]
- Fernández-Cebrián R, Araguas RM, Sanz N, García-Marín JL. 2014. Genetic risks of supplementing trout populations with native stocks: a simulation case study from current Pyrenean populations. Can J Fish Aquat Sci 71: 1243–1255. [CrossRef] [Google Scholar]
- Frankham R. 2008. Genetic adaptation to captivity in species conservation programs. Mol Ecol 17: 325–333. [CrossRef] [PubMed] [Google Scholar]
- García-Marín JL, Jorde PE, Ryman N, Utter F, Pla C. 1991. Management implications of genetic differentiation between native and hatchery populations of brown trout (Salmo trutta) in Spain. Aquaculture 95: 235–249. [CrossRef] [Google Scholar]
- Goudet J, Jombart T. 2022. hierfstat: Estimation and Tests of Hierarchical F-Statistics. R package version 0.5-11, https://CRAN.R-project.org/package=hierfstat [Google Scholar]
- Hamilton KE, Ferguson A, Taggart JB, Tomasson T, Walker A, Fahy E. 1989. Post‐glacial colonization of brown trout, Salmo trutta L.: Ldh‐5 as a phylogeographic marker locus. J Fish Biol 35: 651–664. [CrossRef] [Google Scholar]
- Hammer Ø, Harper DA, Ryan PD. 2001. PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4: 9. [Google Scholar]
- Hansen MM, Ruzzante DE, Nielsen EE, Mensberg KD. 2000. Microsatellite and mitochondrial DNA polymorphism reveals life-history dependent interbreeding between hatchery trout and wild brown trout (Salmo trutta L.). Mol Ecol 9: 583–594. [CrossRef] [PubMed] [Google Scholar]
- Hansen MM, Kenchington E, Nielsen EE. 2001. Assigning individual fish to populations using microsatellite DNA markers. Fish Fish 2: 93–112. [CrossRef] [Google Scholar]
- Hansen MM, Jensen LF. 2005. Sibship within samples of brown trout (Salmo trutta) and implications for supportive breeding. Conserv Genet 6: 297–305. [CrossRef] [Google Scholar]
- Hashemzadeh Segherloo I, Freyhof J, Berrebi P, Ferchaud A-L., Geiger M, Laroche J, Levin BA, Normandeau E, Bernatchez L. 2021. A genomic perspective on an old question: Salmo trouts or Salmo trutta (Teleostei: Salmonidae)? Mol Phylogenet Evol 162: 107204. [CrossRef] [PubMed] [Google Scholar]
- Henry T, Ferguson A. 1985. Kinetic studies on the lactate dehydrogenase (LDH-5) isozymes of brown trout, Salmo trutta L. Comp Biochem Physiol B 82: 95–98. [CrossRef] [PubMed] [Google Scholar]
- Horreo JL, Abad D, Dopico E, Oberlin M, García-Vázquez E. 2015. Expansion of non-native brown trout in South Europe may be inadvertently driven by stocking: molecular and social survey in the North Iberian Narcea River. Int J Mol Sci 16: 15546–15559. [CrossRef] [PubMed] [Google Scholar]
- Jadan M, Strunjak‐Perović I, Topić Popović N, Čož‐Rakovac R. 2015. Three major phylogenetic lineages of brown trout (Salmo trutta Linnaeus, 1758) in the Krka River system (Croatia) revealed by complete mitochondrial DNA control region sequencing. J Appl Ichthyol 31: 192. [CrossRef] [Google Scholar]
- Jones AG, Small CM, Paczolt KA, Ratterman NL. 2009. A practical guide to methods of parentage analysis. Mol Ecol Resour 10: 6–30. [Google Scholar]
- Jones OR, Wang J. 2010. COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol Ecol Resour 10: 551–555. [CrossRef] [Google Scholar]
- Jug T, Berrebi P, Snoj A. 2005. Distribution of non-native trout in Slovenia and their introgression with native trout populations as observed through microsatellite DNA analysis. Biol Conserv 123: 381–388. [CrossRef] [Google Scholar]
- Kanjuh T. 2023. Genetic diversity of brown trout (Salmo trutta L., 1758) of the Danube basin on the territory of Croatia. University of Zagreb, University of Belgrade. International dual doctorate (Cotutelle). [Google Scholar]
- Laikre L, Antunes A, Apostolidis A, Berrebi P, Dugid A, Ferguson A, García-Marín JL, Guyomard R, Hansen MM, Hindar K, Koljonen ML, Largiader C, Martínez P, Nielsen EE, Palm S, Ruzzante DE, Ryman N, Trianthaphyllidis C. 1999. Conservation genetic management of brown trout (Salmo trutta) in Europe. Report by the concerted action on identification, management and exploitation of genetic resources in the brown trout (Salmo trutta), » TROUTCONCERT»; EU FAIR CT97–3882. Silkeborg, Danmarks fiskeri undersrgelser, p 91. [Google Scholar]
- Leigh JW, Bryant D. 2015. PopART: Full‐feature software for haplotype network construction. Methods Ecol Evol 6: 1110–1116. [CrossRef] [Google Scholar]
- Lenhardt M, Smederevac-Lalić M, Hegediš A, Skorić S, Cvijanović G, Višnjić-Jeftić Ž, Djikanović V, Jovičić K, Jaćimović M, Jarić I. 2020. Human impacts on fish Fauna in the Danube River in Serbia: current status and ecological implications. In: Human Impact on Danube Watershed Biodiversity in the XXI Century. Cham: Springer, pp. 257–279. [CrossRef] [Google Scholar]
- Lerceteau-Köhler E, Weiss S. 2006. Development of a multiplex PCR microsatellite assay in brown trout Salmo trutta, and its potential application for the genus. Aquaculture 258: 641–645. [CrossRef] [Google Scholar]
- Lewis PO, Zaykin D. 2001. Genetic data analysis: computer program for the analysis of allelic data. Version 1.0 (d16c). http://www.softsea.com/download/GDA-Genetic-Data-Analysis.html [Google Scholar]
- Marić S, Sušnik S, Simonović P, Snoj A. 2006. Phylogeographic study of brown trout from Serbia, based on mitochondrial DNA control region analysis. Genet Sel Evol 38: 1–20. [CrossRef] [Google Scholar]
- Marić S, Simonović P, Razpet A. 2010. Genetic characterization of broodstock brown trout from Bled fish-farm, Slovenia. Period Biol 112: 145–148. [Google Scholar]
- Marić S, Sušnik Bajec S, Schöffmann J, Kostov V, Snoj A. 2017. Phylogeography of stream-dwelling trout in the Republic of Macedonia and a molecular genetic basis for revision of the taxonomy proposed by S. Karaman. Hydrobiologia 785: 249–260. [Google Scholar]
- Marić S, Stanković D, Sušnik Bajec S, Vukić J, Šanda R, Stefanov T, Nikolić D, Snoj A. 2022. Perils of brown trout (Salmo spp.) mitigation-driven translocations: a case study from the Vlasina Plateau, Southeast Serbia. Biol Invasions 24: 999–1016. [CrossRef] [Google Scholar]
- McLean JE, Seamons TR, Dauer MB, Bentzen P, Quinn TP. 2008. Variation in reproductive success and effective number of breeders in a hatchery population of steelhead trout (Oncorhynchus mykiss): examination by microsatellite-based parentage analysis. Conserv Genet 9: 295–304. [CrossRef] [Google Scholar]
- McMeel OM, Hoey EM, Ferguson A. 2001. Partial nucleotide sequences, and routine typing by polymerase chain reaction-restriction fragment length polymorphism, of the brown trout (Salmo trutta) lactate dehydorgenase, LDHC1*90 and *100 alleles. Mol Ecol 10: 29–34. [CrossRef] [PubMed] [Google Scholar]
- Poteaux C, Bonhomme F, Berrebi P. 1999. Microsatellite polymorphism and genetic impact of restocking in Mediterranean brown trout (Salmo trutta L). Heredity 82: 645–653. [CrossRef] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [CrossRef] [PubMed] [Google Scholar]
- Pustovrh G, Snoj A, Sušnik Bajec S. 2014. Molecular phylogeny of Salmo of the western Balkans, based upon multiple nuclear loci. Genet Sel Evol 46: 1–12. [CrossRef] [Google Scholar]
- Righi T, Fasola E, Iaia M, Stefani F, Volta P. 2023. Limited contribution of hatchery-produced individuals to the sustainment of wild marble trout (Salmo marmoratus Cuvier, 1829) in an Alpine basin. Sci Total Environ 892: 164555. [CrossRef] [PubMed] [Google Scholar]
- Rousset F. 2008. Genepop'007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8: 103–106. [CrossRef] [PubMed] [Google Scholar]
- R Core Team. 2022. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing. https://www.R-project.org/. [Google Scholar]
- Sanz N, Araguas RM, Fernandez R, Vera M, García-Marín JL. 2009. Efficiency of markers and methods for detecting hybrids and introgression in stocked populations.Conserv Genet 10: 225–236. [CrossRef] [Google Scholar]
- Sanz N. 2018. Phylogeographic history of brown trout: a review. In Lobón-Cerviá J, Sanz N, eds. Brown Trout: Biology, Ecology and Management New York: Wiley, pp. 17–63. [Google Scholar]
- Saura M, Faria R. 2011. Genetic tools for restoration of fish populations. J Appl Ichthyol 27: 5–15. [CrossRef] [Google Scholar]
- Sayers EW, Cavanaugh M, Clark K, Pruitt KD, Schoch CL, Sherry ST, Karsch-Mizrachi I. 2022. GenBank. Nucleic Acids Res 50: D161–D164. [CrossRef] [PubMed] [Google Scholar]
- Schmidt T, Zagars M, Roze A, Schulz R. 2017. Genetic diversity of a Daugava basin brown trout (Salmo trutta) brood stock. Knowl Manag Aquat Ecosyst 418: 55. [CrossRef] [EDP Sciences] [Google Scholar]
- Simonović P, Tošić A, Škraba Jurlina D, Nikolić V, Piria M, Tomljanović T, Šprem N, Mrdak D, Milošević D, Bećiraj A, Dekić R, Povž M. 2017. Diversity of brown trout Salmo cf. trutta in the River Danube basin of western Balkans as assessed from the structure of their mitochondrial control region haplotypes. J Ichthyol 57: 603–616. [CrossRef] [Google Scholar]
- Snoj A, Bravničar J, Marić S, Sušnik Bajec S, Benaissa H, Schöffmann J. 2021. Nuclear DNA reveals multiple waves of colonisation, reticulate evolution and a large impact of stocking on trout in north-west Africa. Hydrobiologia 848: 3389–3405. [Google Scholar]
- Snoj A, Marić S, Sušnik Bajec S, Berrebi P, Janjani S, Schoffmann J. 2011. Phylogeographic structure and demographic patterns of brown trout in North-West Africa. Mol Phylogenet Evol 61: 203–211. [CrossRef] [PubMed] [Google Scholar]
- Splendiani A, Ruggeri P, Giovannotti M, Pesaresi S, Occhipinti G, Fioravanti T, Lorenzoni M, Nisi Cerioni P, Caputo Barruchi V. 2016. Alien brown trout invasion of the Italian peninsula: the role of geological, climate and anthropogenic factors. Biol Invasions 18: 2029–2044. [CrossRef] [Google Scholar]
- Szpiech ZA, Jakobsson M, Rosenberg NA. 2008. ADZE: a rarefaction approach for counting alleles private to combinations of populations. Bioinformatics 24: 2498–2504. [CrossRef] [PubMed] [Google Scholar]
- Škraba Jurlina D, Marić A, Karanović J, Nikolić V, Brkušanin M, Kanjuh, T, Mrdak D, Simonović P. 2018. Effect of the introgression of Atlantic brown trout, Salmo trutta, into Adriatic trout, Salmo farioides in a stream at the drainage area of the Adriatic Sea basin of Montenegro. Acta Ichthyol Piscat 48: 363–372. [CrossRef] [Google Scholar]
- Škraba Jurlina D, Marić A, Mrdak D, Kanjuh T, Špelić I, Nikolić V, Piria M, Simonović P. 2020. Alternative life-history in native trout (Salmo spp.) suppresses the invasive effect of alien trout strains introduced into streams in the western part of the Balkans. Front Ecol Evol 8: 188. [CrossRef] [Google Scholar]
- Tamura K, Nei M. 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol 10: 512–526. [PubMed] [Google Scholar]
- Tošić A, Škraba D, Nikolić V, Čanak Atlagić J, Mrdak D, Simonović P. 2016. Haplotype diversity of brown trout Salmo trutta (L.) in the broader Iron Gate area. Turk J Zool 40: 655–662. [CrossRef] [Google Scholar]
- Uiblein F, Jagsch A, Honsig‐Erlenburg W, Weiss S. 2001. Status, habitat use, and vulnerability of the European grayling in Austrian waters. J Fish Biol 59: 223–247. [Google Scholar]
- Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P. 2004. MICRO‐CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538. [CrossRef] [Google Scholar]
- Veličković T. 2023. Koncipiranje modela za održivo korišćenje populacija kompleksa potočne pastrmke (Salmo spp.) na području Srbije. University of Kragujevac: Doctoral Dissertation. [Google Scholar]
- Veličković T, Snoj A, Simić V, Šanda R, Vukić J, Barcytė D, Stanković D, Marić S. 2023. A new perspective on the molecular dating of the brown trout complex with an extended phylogeographic information on the species in Serbia. Contrib Zool 92: 362–389. [Google Scholar]
- Vera M, Cortey M, Sanz N, García‐Marín JL. 2010. Maintenance of an endemic lineage of brown trout (Salmo trutta) within the Duero river basin. J Zoolog Syst Evol Res 48: 181–187. [CrossRef] [Google Scholar]
- Vera M, Aparicio, E, Heras S, Abras A, Casanova A, Roldán M-I., García-Marin J-L. 2023. Regional environmental and climatic concerns on preserving native gene pools of a least concern species: brown trout lineages in Mediterranean streams. Sci Total Environ 862: 160739. [CrossRef] [PubMed] [Google Scholar]
- Wang C, Schroeder KB, Rosenberg NA. 2012. A maximum-likelihood method to correct for allelic dropout in microsatellite data with no replicate genotypes. Genetics 192: 651–669. [CrossRef] [PubMed] [Google Scholar]
- Ward RD. 2000. Genetics in fisheries management. Hydrobiologia 420: 191–201. [Google Scholar]
- Weir BS, Cockerham CC. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38: 1358. [Google Scholar]
- Williams SE, Hoffman EA. 2009. Minimizing genetic adaptation in captive breeding programs: a review. Biol Conserv 142: 2388–2400. [CrossRef] [Google Scholar]
Cite this article as: Veličković T, Snoj A, Bravničar J, Simić V, Šanda R, Vukić J, Barcytė D, Stanković D, Marić S. 2024. Population-genetics analysis of the brown trout broodstock in the “Panjica” hatchery (Serbia) and its conservation applications. Knowl. Manag. Aquat. Ecosyst., 425. 19
All Tables
Sampling sites, number of individuals sampled (N), drainage/origin, and geographic coordinates of Panjica hatchery and reference populations. Panjica hatchery groups: (Group 1) native stock, (Group 2) admixed stock, (Group 3) introduced stock.
Frequency of mtDNA CR haplotypes, LDH alleles and genotypes, and LDH and microsatellite DNA genetic diversity of Panjica hatchery broodstock.
Frequency of mtDNA haplotypes, LDH genotypes, and microsatellite DNA gene diversity for three groups of Panjica hatchery broodstock and reference populations. Panjica hatchery groups: (Group 1) native stock characterized by DA mtDNA CR haplotype, 100/100 genotype and a very high proportion of DA alleles (> 0.99), (Group 2) admixed stock where discrepancies on different markers were detected, (Group 3) introduced stock characterized by AT mtDNA CR haplotype, 90/90 genotype and a very high proportion of AT alleles (> 0.99).
Pairwise ФST (mtDNa CR; above) and FST values (nuclear loci – combined microsatellites and LDH gene; bellow) with significance values, offsprings of the novel broodstock where genotyped at mtDNA CR using only RFLP approach that discriminates between DA and AT lineage.
All Figures
|
Fig. 1 Median-joining network of brown trout mtDNA CR haplotypes. Previously described haplotypes that were not detected in this study are represented by grey circles of the same size. For haplotypes detected in the Panjica hatchery broodstock, the size of the cirlces reflects the frequency of each haplotype, and they are color coded (red – DA, blue – AT lineage). Mutations are represented by slashes crossed with the network branches. The black circle indicates an extinct, ancestral or unsampled haplotype. |
| In the text | |
|
Fig. 2 Estimated population structure as inferred by STRUCTURE analysis of microsatellite DNA data. White lines separate sampling sites. The most likely K for the analyzed samples is based on the ΔK method; no further structures were detected after the first step and within the excluded clusters. The population structure of the Panjica hatchery broodstock is magnified and shown between the arrows, with each column representing one individual (the number corresponds to the nominal number of individuals from Tab. S4). The red color illustrates the genetic proportion of the DA and the blue of the AT. |
| In the text | |
|
Fig. 3 mtDNA haplotype, LDH genotype, and proportion of Danubian alleles of microsatellite DNA of each individual from the Panjica hatchery broodstock including the novel broodstock. The red color illustrates the genetic proportion of the DA and the blue of the AT. Each column represents one individual on three different markers (the number of individuals corresponds to the nominal number from Tab. S4). The group assigned to the individual corresponds to Table S4. |
| In the text | |
|
Fig. 4 NewHybrids analyses using uniform priors based on microsatellite data. The color illustrates the estimated posterior probabilities that an individual belongs to one genotype frequency categories: DA lineage (red), AT lineage (blue), F1 hybrids (pink), F2 hybrids (violet), backcrosses with DA (turquoise), and crosses with AT (orange). |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.